Expedited regulatory pathways: A toolbox to provide innovative medicines to patients
"The revision of the pharmaceutical legislation is a once in a generation opportunity. Leveraging this opportunity to enhance and future proof expedited regulatory pathways is essential to ensure the European regulatory framework can continue to support the development and regulatory approval of innovative medicines."
Nadège Le Roux, Senior Director Regulatory Policy & Intelligence at Bristol Myers Squibb
Introduction
Evidence generation for innovative medicines requires a global approach, both now and in the future, to meet regulatory requirements around the world. Evidence generation also continues long after the first registration of a new medicine. Some medicines benefit from increased support, such as enhanced regulator interactions, due to the level of complexity in development or unmet medical need and/or the ethical urgency with which a treatment should be integrated into best standard of care. Expedited regulatory pathways (ERPs)[1] were developed by multiple international authorities to respond to these needs, creating enhanced early and frequent interactions to support and facilitate innovative treatment development and rapid processes to increase efficiency and reduce time both in development and subsequent regulator assessment. ERPs can be considered a ‘regulatory toolbox’ that can be leveraged to best support the development and efficient assessment of important innovative treatments, so patients can receive them in a timely manner.
In recent years, ERPs have expanded worldwide (Real-time Oncology Review ‘RTOR’ and Regenerative medicine-advanced Therapy Designation ‘RMAT’ in US, China, Australia…) to better support development and improve the speed and efficiency of regulator review of new technologies. Furthermore, ERPs adding work-sharing processes or collaborative platforms (e.g. Innovative Licensing and Access Pathway ‘ILAP’ with project ORBIS) can deliver faster access to new therapies by remaining in the global first wave to innovation while helping to manage resource capacity and expertise[2]. EFPIA welcomes the ongoing reflexion on a future-proofed regulatory framework for ERP, and beyond[3].
The development, submission and/or regulatory review of innovative medicines is accelerated by ERPs, but use of this regulatory toolbox in Europe is limited compared to other regions. Consequently, Europe’s ability to approve innovative medicines efficiently and rapidly to the market is reduced, resulting in delayed access for European patients. ERPs are a necessity for a future-proofed and viable regulatory framework that European patients can rely on for continued delivery of key treatment innovation. Importantly, ERPs are anchored on the same standards for treatment development with regards to safety, quality and efficacy. To ensure European patients continue to benefit from treatment innovations, the ‘toolbox’ needs to be reviewed, and our key proposals are outlined below.
Current situation
The current toolbox of European ERPs is used infrequently compared to similar international pathways. EMA had the second lowest percentage (37%) of medicines approved through an expedited review in comparison to five other major authorities in 2020[4]. The FDA had the highest percentage of new medicines approved via expedited pathways (74%) followed by Swissmedic (61%), and Therapeutic Goods Administration (TGA) in Australia (56%). Moreover, the complexity of the EU regulatory ecosystem, organised in two layers (e.g. National and EMA/EU one), slows the potential of ERP tools.
The flexibility, agility and possibility of combining various ERPs in the EU needs to be enhanced. Some member states are hesitant about ERPs as they imply a more iterative evidence generation with a first registration based on a first dossier responding to the core benefit/risk that may not yet reflect the long-term need decisions of other downstream decision-makers. Health Technology Assessment and Pricing & Reimbursement authorities must focus on how to support delivery of treatments to patients who need them by engaging in a conversation on evidence generation, including generation beyond the first registration to further characterise the treatment and support access decisions. Important conversations on skills and resources are taking place currently within the EU Network and we would like to re-iterate the EFPIA position that the innovative industry is willing to pay more for the additional support provided by some key ERP components: iterative scientific advice, PRIME and dynamic regulatory assessment[5].
Future proofing the ERP Toolbox
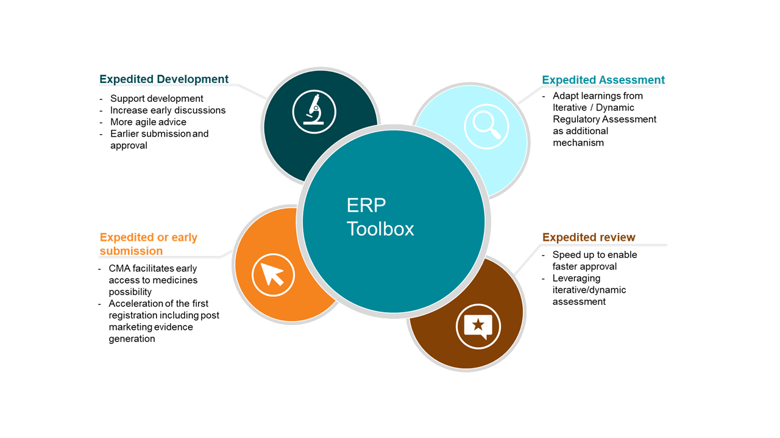
ERP within the toolbox should be used flexibly to suit specific situations. ERPs include the following tools and methods that are not mutually exclusive and can be used on their own or combined to adapt to the context and support efficiently the expedited development and efficient treatment approval:
- Expedited development:
- To actively support the development of ground-breaking medicines with enhanced and active iterative dialogue to align on appropriate evidence generation, needed for regulatory assessment and future decisions (e.g. for PRIME designated medicines).
- Facilitate early multistakeholder discussions across the EU network (EMA and national competent authorities) and other stakeholders as needed.
- Where appropriate, more agile and rapid scientific advice (SA) to efficiently support development.
- Where and when appropriate, earlier submission and approval with a dataset based at an agreed time point (e.g., surrogate endpoints, phase 2 data only), with robust commitments to continue with post licensing evidence generation (e.g. in context of Conditional Marketing Authorisation ‘CMA’). A first time to registration is to support patient needs while the evidence generation is pursuing its pace to build the knowledge of the innovative medicine.
- Expedited assessment (iterative/dynamic regulatory assessment):
- Rolling reviews were successfully used during the COVID pandemic, leading to greatly reduced development and authorisation timelines. Although this level of resource can only be released in rare public health emergencies, there are learnings that must be extracted from iterative release and assessment of data (also called Dynamic Regulatory Assessment ‘DRA’) and applied more broadly. DRA should be thoughtfully applied to ERP as an additional mechanism to support development and efficient regulatory approval, and we recommend beginning with a pilot of this concept as an action connected to the PRIME 5-year review[6],[7], although the concept applies more broadly (see below). In a close future IT tools and digitisation will facilitate smooth processes and connectedness that ERPs greatly need.
- Expedited review:
- Regulatory authorities can speed up the review of certain products to enable faster approval with the ERP called Accelerated Assessment (AA).
- Leveraging iterative/ dynamic regulatory assessment could be helpful in this setting thanks to a stagged approach. A pilot would help to discuss this in a concrete way.
- Expedited or early submission:
- Evidence generation is a continuum and continues long after the first registration of a new medicine. CMA can facilitate early access to medicines, as seen in for e.g. COVID therapeutics/vaccines. In CMA, the benefit/risk of earlier regulatory approval is assessed taking into account the context of e.g. a seriously debilitating or life-threatening disease, public health conditions and the remaining uncertainty with regards to the ability to generate the required data. Part of the first approval is decided with a set of pre-approved commitments to subsequently generate the additional data needed towards a full marketing authorisation. Thoughtful evidence generation planning can support timely assessment for HTA/pricing reimbursement. The continuum knowledge on a product is a reality that each stakeholder of the EU ecosystem should embrace.
- ERP allows acceleration of the first registration, while evidence generated actively and rapidly continue to complement and confirm the initial knowledge of the product. It is important to state that post marketing evidence generation always continues in this setting and always in agreement with regulators.
Going forward
As recorded in the European Pharmaceutical strategy, PRIME ought to be incorporated in the regulatory framework to fully optimise the support and acceleration of innovative medicines being launched on the EU market. This tool will also be improved and invested in according to the EMA Regulatory science and HMA European Medicines Agencies Network Strategy to 2025. It will be crucial to align on the eligibility criteria for PRIME. For example a too narrow definition of ‘Unmet Medical Need’ would further limit the use of this tool for patients’ faster access and EFPIA stands firm that unmet medical need has to take into account patient, societal and health system perspectives[8]. Furthermore, rolling review is currently only being used to manage health emergencies like the COVID pandemic[9]. Learnings from rolling review and stakeholder experiences must be thoughtfully applied to a broader set of medicines, as has happened in similar countries and regions. A crucial component for effective ERP evolution is the implementation of effective knowledge management and institutional memory within the EMA and the EU Network (at large) to connect all activities within the EU Network.
The COVID pandemic has shown an urgent need for a fast approach to addressing unmet medical need during a health emergency and has given a glimpse of the possibilities European patients have been anticipating for other life-threatening diseases and conditions. For Europe to stand its ground amongst other international regulatory authorities, in an era of innovation and global development, it is crucial to evolve and future proof EU ERPs. We believe this evolution can be done efficiently, without compromising on the safety, efficacy and quality of future medicines. Furthermore, we strongly need support from national competent authorities as we future-proof the EU’s regulatory system to reclaim competitive advantage and continue delivering the treatments patients need now, and in future. The effectiveness of the EU system will need improved allocation of local and national resources, setting clear priorities for innovation in the EU network. These reflections ought to be addressed urgently if the EU wants to support and deliver innovative medicines within similar timeframe as other Global agencies to be part of the first global wave to innovation. In parallel, an important conversation is needed to optimize fees, expertise and resourcing capacity generally within the EU network as this underpins functioning of any future system[10].
[1] In the European Union, ERPs include PRIME, Accelerated Assessment and Conditional Marketing Authorisation.
[2] https://cirsci.org/wp-content/uploads/dlm_uploads/2022/06/CIRS-RD-Briefing-85-NAS-list-v2.2.pdf
[3] ERPs include in the US 2 new tools e.g. RTOR and RMAT- in China, in addition to priority and special review pathway, NMPA introduced Breakthrough and Conditional Approval in 2020 to accelerate development and approval of drugs with significant clinical value. In Australia, in early 2022 TGA consulted on new Priority review pathway for biologicals including human cell and tissue therapies.
[4] https://cirsci.org/wp-content/uploads/dlm_uploads/2021/06/CIRS-RD-Briefing-81-6-agencies-v5.pdf
[5] https://www.clinicaltherapeutics.com/article/S0149-2918(21)00456-2/fulltext
[6] https://cirsci.org/wp-content/uploads/dlm_uploads/2021/06/CIRS-RD-Briefing-81-6-agencies-v5.pdf
[7] https://www.ema.europa.eu/en/documents/report/prime-5-years-experience_en.pdf
[8] https://www.efpia.eu/about-medicines/development-of-medicines/unmet-medical-need/
[9] https://pubmed.ncbi.nlm.nih.gov/35123802/
[10] https://efpia.eu/media/636798/efpia-regulatoryroadtoinnovation_triptych_v07-final-neworder_pbp.pdf

