Regulations, safety & supply
Safer, better, faster.
Our well-functioning regulatory system also plays a significant role in enhancing Europe's competitiveness in the global race of attracting investments in the life sciences sector. This is especially important given the fierce competition we face from other global economies, particularly the United States and China.
Number of new chemical or biological entities
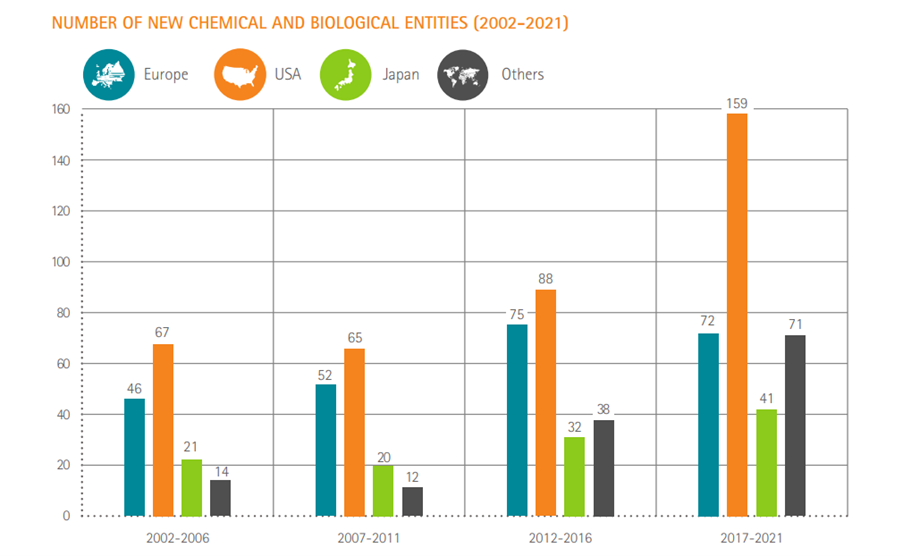
Source: SCRIP – EFPIA calculations (according to nationality of mother company) – link
With the revision of the EU pharmaceutical legislation, we are now in an important moment for Europe’s ambition to be a global leader in innovation.
Regulatory science is a field looking into developing new tools, standards and approaches to evaluate the efficacy, safety, quality and performance of vaccines and treatments, in order to facilitate a sound and transparent regulatory decision-making. It can be a fast-moving and exciting area, and is transforming the way new therapeutics are developed through advances in science, data analytics, the ability to collect real-world data and turn them into real-world evidence.

The total number of Next-Generation Biotherapeutics (NGBs) – defined as cell, gene and nucleotide therapies – in the development pipeline went from 120 in 2015 to 269 by the end of 2018.
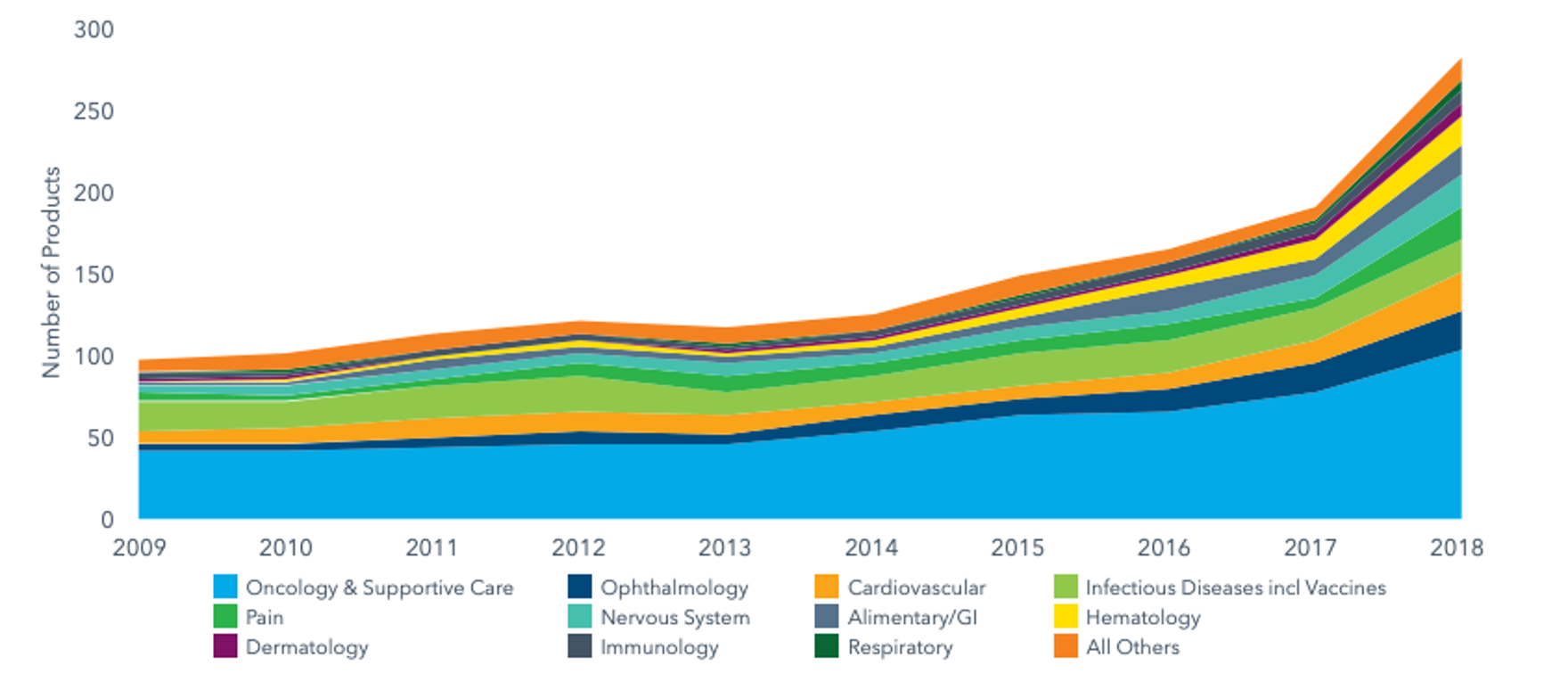
NGBs make around 10% of the total late-stage R&D pipeline and have more than doubled in number over the past three years as new pathways for disease treatment and cure.
Source: IQVIA, The Changing Landscape of Research and Development, April 2019, link
Working together to deliver more for patients, in Europe and beyond
|
EFPIA and its members provide their practical experience and improvement suggestions on all phases of medicines development, manufacturing and regulatory aspects. All of EFPIA’s submissions to regulatory consultations are made available online and you can access them here. |
The framework also covers the provision of evidence that medicines are researched and manufactured in compliance with regulations (Good Laboratory/Clinical/Manufacturing Practices), and the continuous monitoring of medicines once they have entered the market in order to detect potential safety and quality issues. An important role the framework plays is around relaying information and informing the development and decision making across medicine’s lifecycle - including real-world data collected in patients’ Health Records or registries.
As part of the medicine’s licence, environmental and sustainable manufacturing issues need to be addressed as well, and compliance with the respective legislative requirements needs to be proven. Furthermore, there is always a need for a revision or an update to the marketing authorisation details at some point, such as routine continuous updates to the existing licence. New scientific information could also lead to an extension of application to be able to give the medicine to additional patients’ groups, such as children, pregnant women and elderly.
Upon regulatory approval, there is a separate evaluation and a decision (Health Technology Assessment) on price, as well as negotiations of potential reimbursement conditions covered by national healthcare systems.
Looking beyond Europe
|
EFPIA and the European Commission/European Medicines Agency are also founding members of The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). ICH's mission is to achieve greater harmonisation worldwide to ensure that safe, effective, and high-quality medicines are developed and registered in the most resource-efficient manner. You can find more information on ICH via this link. |
Global regulatory convergence is a means of aligning regulatory requirements across the world via a process of gradual uptake of internationally recognised technical and procedural standards and principles, with the aim of improving healthcare. It does not necessarily mean that all laws and requirements are harmonised, but that greater regulatory cooperation is made possible. Such approaches cut the so called “regulatory red tape” and allow for faster access by patients to quality, safe and efficient vaccines and treatments all around the globe.



