Regulatory Pathways for an Integral Drug-Device Combination Product Platform based on Quality by Design (Guest Blog)
Established principles of Quality by Design [1] and Risk Management [2], [3], which are commonly practiced in the development of medicinal products, are proposed for integral drug-device combination (iDDC) product development within the concept of a platform device. An iDDC product manufacturer may choose to use the same or very similar device system to contain and administer different medicinal products. Therefore, the product device technical information would be identical or comparable between products and these products may constitute a platform. Using a platform approach in the development of an iDDC product provides an opportunity to streamline the regulatory review procedures for the product by minimising repeat review of the same information by the EMA and Notified Body (NB) and the risk of prolonging the review period to approval and launch of the new iDDC product. As such, a platform approach has the potential to bring innovative iDDC products to patients more quickly.
It is acknowledged that a platform approach would still need to consider the specific medicinal product-device interactions, intended user, uses, and use environment to ensure safety, efficacy and usability of the product in each combination.
The soon to be implemented Article 117 [4] of the Medical Device Regulation requires that an EMA Marketing Authorisation Application (MAA) for an iDDC product includes a Notified Body Opinion (NBOp) to demonstrate compliance with the General Safety and Performance Requirements (GSPRs). The NBOp involves review by a Notified Body (NB) of the technical device and drug-device combination to assess performance, safety and usability.
EFPIA has published a reflection paper on how a platform device approach for a new iDDC product could be developed to allow for a platform NBOp that could be reused whenever the new product lies within the platform parameters (design space) [5]. The platform would be comprised of a set of characterised, verified and validated iDDC configurations. These configurations could be paired with drugs that vary in a defined set of critical performance parameters, e.g. a prefilled pen platform could be paired with different drugs that vary in medicinal product viscosity and volume. The configurations also may allow for use of different spring strengths and/or needle bore, for administration of the various drugs. All of these variable components would be qualified as part of the platform space, such that they can be mixed and matched as appropriate for the particular iDDC medicinal product. The external design is unchanged, and the spring is the only internal design change, in this scenario. These variable parameters form a device design space (see illustration) and with increasing complexity of variables, Design of Experiments (as an example method) could be used to identify the key configurations that require characterisation to define the platform device design space.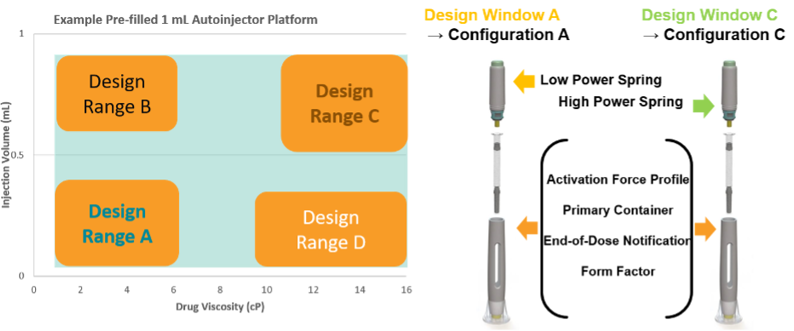
The platform technical dossier would contain all the data to support the General Safety and Performance Requirements (GSPR) for designs A, B, C and D. Any new prefilled pen of this same basic design (Form Factor) used with a drug of a different viscosity and/or volume that lies within the characterised ranges of the bracketed parameters would be considered to lie within the platform - and the platform NBOp applies. The platform data in the dossier would include testing using a surrogate solution that is appropriate and justified for use in tests for the GSPR parameters investigated. The user/patient population would be a defined criterion to be considered in developing the platform design space. The ‘use’ (indications) should be appropriately generic for general purpose use e.g. subcutaneous injection – for the stated patient population and consider the design space in terms of chronic, acute, emergency use etc.
On this basis, a platform NBOp is considered appropriate.
The MAA would include: (i) a complete module 3 of the Common Technical Document (CTD) with all the medicinal product information, including product compatibility with the device constituent parts, (ii) the platform NBOp and justification of the applicability of the NBOp, and (iii) a risk assessment to identify and mitigate any gaps in knowledge of the iDDC product.
The platform iDDC product regulatory review would greatly benefit from a clear division of responsibility for assessment of drug-device compatibility data (medicinal product-device interactions). Since iDDC products are regulated as medicinal products, EFPIA suggests that it is the remit of the EMA to review compatibility data that pertain to the drug and drug-device interaction, including the device performance critical parameters using the specific medicinal product. The EMA review of the CTD would include the relevant product-specific drug-device compatibility data. The NB should review the device specific data and performance, including Usability Studies, which can be satisfactorily evaluated using surrogate solutions
The platform NBOp is considered particularly suitable when the iDDC product manufacturer has designed a device platform at first intent. However, it should also be possible to use the NBOp for one product (Product A) for another product (Product B) when the GSPRs are satisfied for both products. Similarly, the Product B CTD would justify use of the Product A NBOp and include a risk assessment and mitigations for any identified gaps. In essence, the data for Product A is a surrogate for Product B.
The similarity of this platform approach to CE-marking is striking. Medical devices marketed in a manner that is not integral to a medicinal product are CE-marked and developed for general use using surrogate solutions. These CE-mark product certificates are reused for different medicinal products. The CE-mark process has a clear distinction of review responsibility lying with the NB since the data pertain to the device and device performance and safety using surrogate solutions. Since CE-marked devices are considered generally safe and suitable for use with different products, then in parallel, products covered by a platform NBOp should also be considered suitable for patient use with different products within the platform space.
Platform devices are being developed and hence there is an urgency for the EC, EMA, NB and Industry to maintain dialogue and find solutions to the review of platform iDDC products and the platform NBOp for an efficient, streamlined process to approved new products of high quality that meet patient needs in a timely manner.
[1] ICH Guideline Q8 (R2) on Pharmaceutical Development (2017), EMA/CHMP/ICH/167068/2004
[2] ICH Guideline Q9 on Quality Risk Management (2015), EMA/CHMP/ICH/24235/2006
[3] Risk Management Guidance for Combination Products (2020), AAMI TIR 105:2020
[4] Article 117; Amendment to Directive 2001/83/EC (2017); Official Journal of the European Union, L 117/88
[5] EFPIA Reflection Paper on Integral Drug-device Combination Product Platform Approach (2021). http://efpia.eu/media/589835/efpia-rp-platform-devices_final16-april-2021.pdf